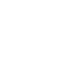![]()


英文标题:Integration of omic networks in a developmental atlas of maize
中文标题:利用整合的组学网络重构玉米发育图谱
作者:Justin W. Walley, Ryan C. Sartor, Zhouxin Shen, Robert J. Schmitz, Kevin J. Wu, Mark A. Urich, Joseph R. Nery, Laurie G. Smith, James C. Schnable, Joseph R. Ecker, Steven P. Briggs
第一单位:University of California San Diego
发表杂志:Science, 2016, 353 (6301) :814-818
解读人:韩乃坚博士
摘要
共表达网络和基因调控网络能够系统地预测单个基因功能。为了进行网络重建,作者建立了一个大规模的与玉米发育过程相关的基因表达谱,其中涉及62547个 mRNA,17862个非修饰蛋白,以及拥有31595个磷酸化修饰位点的6227个磷蛋白。研究发现,基于mRNA表达量而构建的共表达网络与基于蛋白表达量的网络存在较大差异,仅有15%的高度互连核心节点在RNA和蛋白网络中是保守的。尽管如此,基于不同类型数据构建的网络仍然能够富集到相似的本体论类别,并且能够有效预测已知的基因调控关系。多组学(mRNA、蛋白质及磷蛋白)的整合分析能够极大地提高基因调控网络的预测能力。
正文
系统地预测基因功能是生物学领域的一个复杂的挑战。目前随着高通量测序技术的成熟以及成本的极速下降,大量的转录组数据被用于构建基因组范围的基因调控网络(GRNs)和共表达网络。然而,mRNA并不能完全代表蛋白丰度,众多研究显示mRNA丰度与蛋白丰度仅存在较弱的正相关。这意味着加入蛋白数据,可能会极大的改善仅基于转录组的基因调控网络质量。因此在本研究中,作者建立了玉米发育过程中23个组织的转录组、蛋白质组和磷蛋白组的数据,探讨多组学合并数据相比于单组学数据,在构建基因调控网络和共表达网络所呈现的优势。
作者首先利用10979个基因重建共表达网络(图1A)。通过斯皮尔曼相关系数,衡量mRNA和mRNA之间,以及蛋白和蛋白之间的相关性。为了直接比较mRNA和蛋白质共表达网络,并编译一个高置信度的共表达数据集,每一个网络被限制只包括相关指数大于0.75的边界。分析结果显示122029个边界在两个网络中是保守的,占总体的6.1%(图1B)。尽管这个边界的重叠区远大于随机期望的0.8%,但是大多数的基因关系具有网络独特性。即使将边界数目扩大到100万,上述指标也未发生显著改变。这一结果与之前的一系列研究相吻合,也即mRNA和蛋白质具有较弱的正相关。
共表达网络的一个关键特征是存在少量但高连接度的核心节点,通常一个节点代表一个基因。核心节点比非核心节点,在网络完整度和有机体存活中具有更重要意义,因此判定这些所谓的核心节点是网络分析的重心之一。结果显示大多数核心节点是mRNA或蛋白质网络所独有,而共同的核心节点仅占15%左右(图1C)。

图1. 共表达网络分析。(A)假设的非定向共表达子网络显示mRNA和蛋白质网络中保守的(实线)和非保守的(虚线)共表达边界。(B)利用维恩图展示边界保守性;(C)已每个节点的边界数目,X轴代表蛋白数,Y轴代表mRNA数。红色点为基于mRNA分析得到的核心节点,蓝色点为基于蛋白分析得到的核心节点,绿色点为共同获得的核心节点。
对共表达网络中的模块进行富集分析,结果显示大多数模块都能显著的富集到一个或多个Mapman类别。mRNA和蛋白质网络模块在类别富集程度方面总体上相似,但是显著富集的类别中的基因实际存在网络独特性(35%存在于蛋白网络,27%存在于mRNA网络,38%为两者共有)(图2)。这些发现表明不同类型数据,预测到的基因相关性和功能也不尽相同。推测造成mRNA和蛋白质共表达网络不一致的原因,主要是mRNA丰度和蛋白质丰度之间有限的相关性;而有限的相关性,可能受mRNA和蛋白质的稳定性、翻译调控以及蛋白转运等因素影响。

图2. 共表达网络模块的类别富集分析。
相比于共表达网络,GRN能够更直接地展示转录因子及其靶基因(图3A),因此为了深入探索玉米发育过程中的基因表达调控模式,作者分别利用mRNA、蛋白质和磷酸肽数据构建了三个基因调控网络(GRN)。为了构建这些网络,转录因子方面选择了2200个可定量的mRNA、545个可定量的蛋白质,以及441个可定量的磷酸肽,而靶基因数据集则来自于41021个定量的mRNA。已公布的两类经典的玉米转录因子(同源盒转录因子KN1和bZIP转录因子Opaque2)用来进行GRN质量评估,评估方法是绘制工作者受试曲线(ROC)和正确率-召回率曲线。尽管两种曲线显示出三个GRNs在总体质量方面较为相似,但细看得分前500位的结果会发现,基于蛋白数据的两个GRNs能够更准确地预测靶基因(图3B):KN1子网络准确预测到108(mRNA),129(蛋白质)和125(磷酸肽)个靶基因,而O2子网络表现类似。此外,研究还发现在准确预测到的靶基因亦存在GRN独特性,且比例不低(KN1有44%;O2有31%)(图4B)。上述结果表明利用不同表达数据进行GRN预测,能够极大的互补。
将GRN分析扩大到所有转录因子后,作者同样发现存在较低的边界保守性,而大多数边界仍为不同GRN所独有——例如边界数为100万时,93%的边界存在于单一的GRN(图3C);而边界数为20万时,数值则增加到96%。这一发现说明转录因子在mRNA、蛋白质和磷酸化层面的不同积累模式,导致了截然不同的GRN预测结果。
为了检测多组学数据的整合分析,是否能够改善单组学分析结果,作者另外构建了四个GRNs,并利用KN1和O2相关数据来评估网络质量(图3)。结果显示基于多组学的GRN预测优于单组学预测。图3D中显示仅用mRNA信息推断转录因子活性,计算的ROC曲线下方面积(AUC)为0.657;当进一步结合蛋白丰度以及磷酸化水平信息后,AUC增加到0.717。这表明如果进行假阳性率为20%的网络预测时,仅凭mRNA数据,真阳性率为40%,而利用多组学整合分析则能提高到50%。图3E也揭示了类似的结果——如果进行精确度为0.021的网络预测时,仅凭mRNA数据,只有16%的真阳性率,而整合分析则能提高到41%。
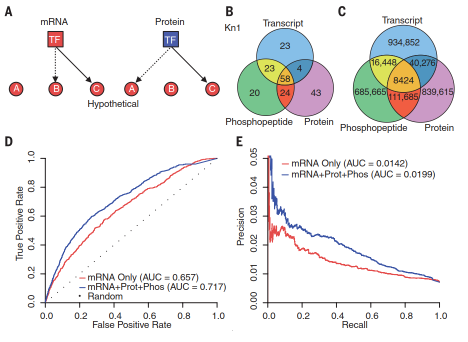
图3. 无监督的基因调控网络(GRN)分析。
提示和启发
1.大多数基因在mRNA水平和蛋白质水平呈现较弱的正相关性,mRNA丰度并不能客观反映蛋白丰度,导致mRNA分析结果的真阳性率相对较低,因此进行蛋白质组学研究是十分必要的。
2.然而由于目前蛋白质组学技术发展相对缓慢,存在通量低、成本高的劣势。如能通过联合其他组学(如转录组、磷蛋白质组、代谢组等)进行整合性分析,则能够获得多样的基因信息,有助于全面系统地解析复杂生物学过程背后的分子调控机制。
3.抗体组是蛋白质组学研究的重要手段之一,不但具有更高的重复性和稳定性,同时还可进行蛋白定位、互作等方面的研究,这一优势是基于质谱技术的蛋白质组学所无法实现的。

艾比玛特医药科技(上海)有限公司
上海市徐汇区桂平路333号聚科生物园区1号楼1-3层
邮箱:market@ab-mart.com
应聘职位:hr@ab-mart.com
订购专线:4006-123-828
销售电话:13916964679(微信同号)
技术支持:15618194176(微信同号)
华南经销商负责(广东,广西,福建,海南):
程经理:手机18616261485(微信同号)
华北经销商负责(北京,天津,河北):
徐经理:手机15618191473(微信同号)
南方经销商负责:
陆经理:手机13122837132(微信同号)
北方及西南经销商负责:
张经理:手机13122150513(微信同号)

微信客服
邮箱:market@ab-mart.com
应聘职位:hr@ab-mart.com
订购专线:4006-123-828
销售电话:13916964679(微信同号)
技术支持:15618194176(微信同号)
华南经销商负责(广东,广西,福建,海南):
程经理:手机18616261485(微信同号)
华北经销商负责(北京,天津,河北):
徐经理:手机15618191473(微信同号)
南方经销商负责:
陆经理:手机13122837132(微信同号)
北方及西南经销商负责:
张经理:手机13122150513(微信同号)
微信客服
沪ICP备17056956号-2 艾比玛特医药科技(上海)有限公司